
What is CAH?
Genetic Causes of Other Rare Forms

Figure 1 shows the normal biosynthetic pathways for mineralocorticoids, glucocorticoids, and adrenal androgens. Each step is catalyzed by a specific enzyme. In Figure 1, the gene coding for these enzymes are indicated in gray. CAH can be caused by a defect in any one of several of these genes.
11ß-Hydroxylase Deficiency
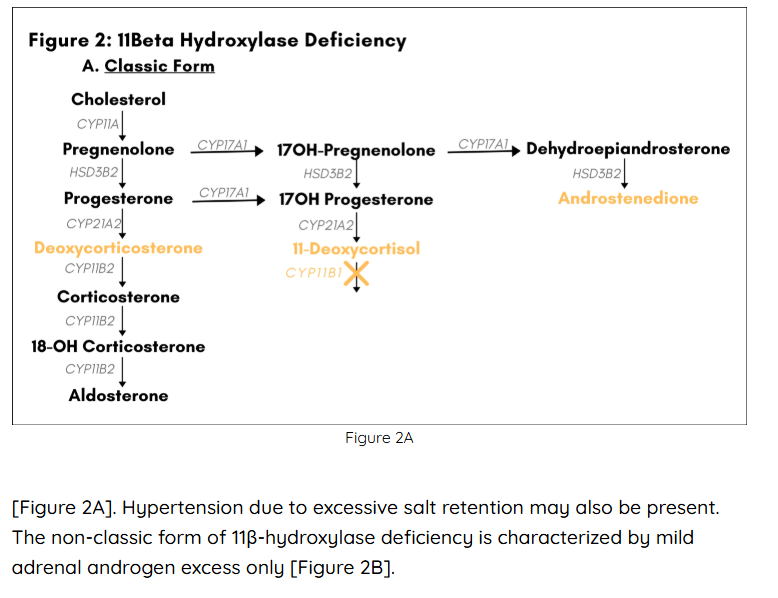
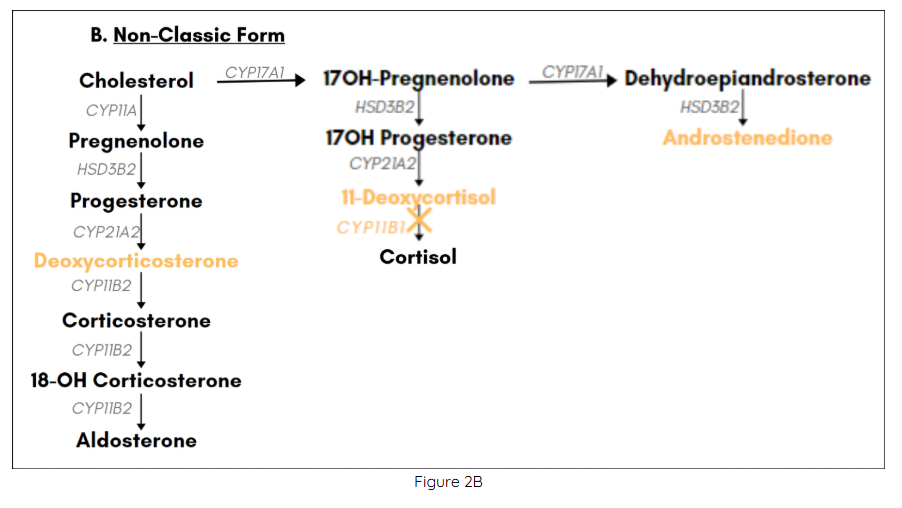
Loss-of-function mutations in the gene CYP11B1 account for about 5-8% of all cases of CAH in Western Europe and the US (4,5). CYP11B1 codes for the cytochrome P-450 enzyme 11β-hydroxylase, which catalyzes the conversion of 11-deoxycortisol to cortisol in the glucocorticoid biosynthetic pathway. Defects in 11β-hydroxylase disrupt glucocorticoid synthesis, and precursors accumulating “upstream” of the enzymatic block are shunted into the biosynthetic pathways for mineralocorticoids and adrenal androgens. Patients are protected from an adrenal crisis by presence of corticosterone, which possesses glucocorticoid activity. However, increased levels of deoxycorticosterone can give rise to hypertension, since this intermediate product in the mineralocorticoid biosynthetic pathway shows weak mineralocorticoid activity. Overproduction of adrenal androgens leads to prenatal and/or postnatal virilization in genetic females and postnatal virilization in genetic males. The degree of virilization depends on the extent to which 11β-hydroxylase activity is impaired, which also determines the degree of excess mineralocorticoid activity. In the classic form of 11β -hydroxylase deficiency, which accounts for about two thirds of all cases, genetic females are born with ambiguous genitalia, and both males and females undergo premature adrenarche if untreated.
17a-Hydroxylase Deficiency

Loss-of-function mutations in the gene CYP17A1 account for about 1% of CAH in most populations, but represent the second most common cause of CAH in Brazil (6). CYP17A1 codes for a cytochrome P-450 enzyme with both 17α-hydroxylase and 17,20-lyase activities, which performs a gatekeeper function for the entry of precursor molecules into the glucocorticoid and adrenal androgen biosynthetic pathways: 17α-hydroxylase activity controls the branch point from the mineralocorticoid to the glucocorticoid biosynthetic pathway, and 17,20-lyase activity regulates the branch point from the glucocorticoid to the adrenal androgen biosynthetic pathway [see Fig. 3]. Defects in CYP17A1 that disrupt both the hydroxylase and the lyase activity affect the synthesis of glucocorticoids and androgens in the adrenal cortex as well as steroid synthesis in the gonads (7). Mineralocorticoid synthesis is not affected, but the block in the entry point to the glucocorticoid biosynthetic pathway leads to an increase in mineralocorticoid precursors such as deoxycorticosterone and corticosterone. This increase in the concentration of corticosterone, which exhibits glucocorticoid activity, compensates for the absence of cortisol and protects patients from an adrenal crisis. Increases in the levels of deoxycorticosterone and corticosterone are also believed to be the cause of the hypertension frequently observed in patients with 17α-hyproxylase deficiency, since both intermediates possess weak mineralocorticoid activity. Disruption of adrenal androgen production and gonadal steroid synthesis causes female or ambiguous external genitalia in 46XY males, sexual infantilism, and primary or, more rarely, secondary amenorrhea in 46XX females.
3ß-Hydroxysteroid Dehydrogenase Deficiency
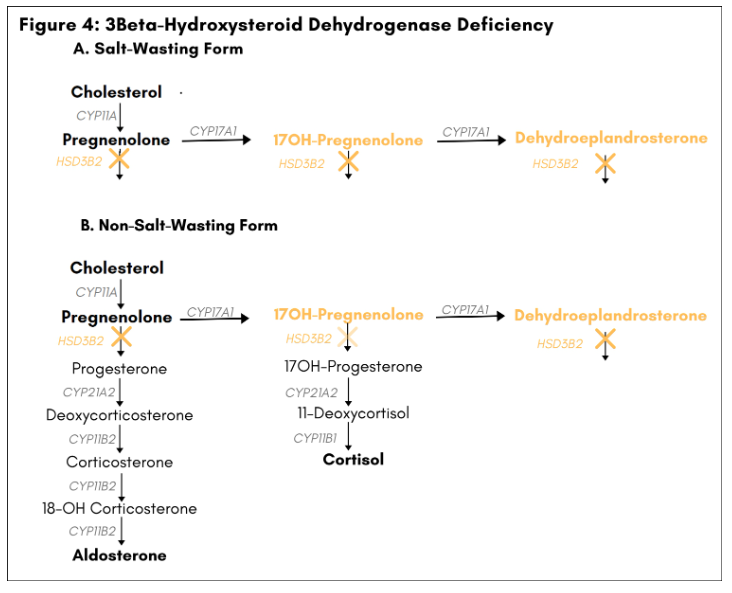
Loss-of-function mutations in the gene HSD3B2 account for about 1% of CAH (8). HSD3B2 codes for the enzyme
3β-hydroxysteroid dehydrogenase (3β-HSD), which catalyzes the conversion of pregnenolone to progesterone in the mineralocorticoid biosynthetic pathway and the conversion of 17OH-pregnenolone to 17OH-progesterone in the glucocorticoid biosynthetic pathway. 3β-Hydroxysteroid dehydrogenase is also necessary for conversion of dehydroepiandrosterone to androstenedione in the adrenals and for the biosynthesis of sex steroids in the gonads. In 46XY males, 3β-HSD deficiency is associated with ambiguous genitalia due to impaired testosterone synthesis in the gonads. In genetic females, 3β-HSD deficiency may lead to premature adrenarche, amenorrhea, or ambiguous genitalia due to conversion of androgen precursors to androgens in peripheral tissues. Severe defects in 3β-HSD activity also cause deficiency in both mineralocortocoids and glucocorticoids and lead to salt wasting and primary adrenal insufficiency in males and females (salt-wasting form). [Figure 4]
Congenital Lipoid Adrenal Hyperplasia
Loss-of-function mutations in the gene STAR, which are associated with congenital lipoid adrenal hyperplasia (lipoid CAH), account for a small percentage of CAH in most populations, but appear to be more common in individuals of Japanese, Korean, or Palestinian ancestry (9). STAR codes for the steroid acute regulatory protein, which fulfills a gatekeeper function for all of steroid biosynthesis by catalyzing the transfer of cholesterol from the cytosol into mitochondria, where the initial steps of steroidogenesis take place. Absence of functional StAR protein reduces cholesterol import into mitochondria by a factor of about ten, leading to impaired biosynthesis of all steroids and accumulation of cholesterol lipid droplets in the cytoplasm of affected cells. However, StAR-protein independent cholesterol import may allow for enough steroid synthesis to prevent acute symptoms in the absence of functional StAR protein, and only the gradual accumulation of lipid droplets leads to complete cessation of all steroidogenesis through generalized damage to the affected cells. For this reason, defects in STAR cause damage to steroidogenic target organs such as the adrenals and the gonads only if they are stimulated to produce steroids. This “two hit” model of lipoid CAH explains why 46XY infants with lipoid CAH are born as phenotypic females, while onset of acute primary adrenal insufficiency and salt wasting may not occur until several weeks or even months after birth. The fetal testis is stimulated and thus damaged by absence of functional StAR early in gestation, leading to lack of testosterone and preventing development of male external genitalia. In the adrenals, in contrast, stimulation prior to birth affects primarily the fetal zone; the definitive zone, which postnatally develops into the zona glomerulosa and zona fasciculata, may therefore remain partially functional for several weeks or months after birth in individuals with reduced StAR-protein activity. Similarly, the ovaries in 46XX individuals remain hormonally silent until puberty, when individual ovarian follicles are stimulated and, in individuals with lipoid CAH, thus damaged during each cycle. However, while 46XX individuals with lipoid CAH can spontaneously enter puberty and may undergo menarche, their cycles remain anovulatory because lack of StAR-protein activity prevents the progesterone surge necessary for ovulation. To learn more about the genetics of CAH, please visit out CAH Overview page.
REFERENCES:
- New MI (2004) An update of congenital adrenal hyperplasia. Ann NY Acd Sci 1038:14-43.
- White PC, Speiser PW (2000) Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocrine Reviews 21:245-291.
- Speiser PW, White PC (2003) Congenital adrenal hyperplasia. N Engl J Med 349:776-88.
- New MI (2003) Inborn errors of adrenal steroidogenesis. Mol Cell Endocrinol 211:75-83.
- Chemaitilly W, Wilson RC, New MI (2003) Hypertension and adrenal disorders. Current Hypertension Reports 5:498-504.
- Costa-Santos M, Kater CE, Auchus RJ, et al (2004) Two prevalent CYP17 mutations and genotype-phenotype correlations in 24 Brazilian patients with 17-hydroxylase deficiency. J Clin Endocrinol Metab 89:49-60.
- Auchus RJ (2001) The genetics, pathophysiology, and management of human deficiencies of P450c17. Endocrinol Metab Clin North Am 30:101-19,vii.
- Simard J, Ricketts M-L, Gingras S, Soucy P, Feltus AF, Melner MH (2005) Molecular biology of the 3beta-hydroxysteroid dehydrogenase/delta5-delta4 isomerase gene family. Endocrine Reviews 26:525-82.
- Miller WL (1997) Congenital lipoid adrenal hyperplasia: the human gene knockout for the steroidogenic acute regulatory protein. J Mol Endocrinol 19:227-40.

CONTACT
2414 Morris Avenue, Ste 110
Union, NJ 07083
Phone: (908) 364-0272
Toll Free: (866) 227-3737
Fax: (908) 686-2019
contact@caresfoundation.org
